




Research Overview
Monitoring cell oxidation with molecular probes
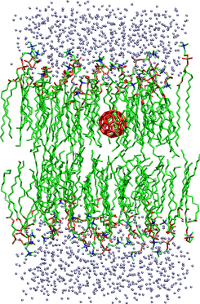
The oxidation of cell membranes has been linked to aging and neurodegenerative diseases, including Alzheimer’s and Parkinson’s disease. The initial steps in the oxidation process are presently an open question, including the role of specific chemical species. Once initiated, the cell membrane undergoes a chain reaction of lipid peroxidation, resulting in a loss of fluidity. We have previously demonstrated that the diffusion kinetics of small molecules and nanoparticles permeating through a membrane is highly dependent on the chemical composition of the permeating particle and the membrane itself. Relatively few definitive facts are known, however, about the driving forces and mechanisms of the passive permeation process. To this end, we are developing an experimental and theoretical framework to rapidly assess the integrity of the membrane in an oxidative environment, see Biophys. J. 99, 144 (2010).
Quantum dynamics
Some photochemical reactions instigate conditions where the dynamics of molecules are influenced by correlated motion of the electrons and atomic nuclei. DNA (nucleic base) molecules, for example, exploit this mechanism to rapidly dissipate absorbed UV radiation that can result in mutation. As such, life as we know it could not exist in the absence of this mechanism. At the atomistic level, this process instigates atomic motions that do not necessarily follow a single (adiabatic) potential energy hypersurface. This breakdown of the Born-Oppenheimer approximation necessitates the refinement of conventional computational approaches used to computationally simulate such reactions. In this set of studies, we apply the mixed quantum-classical, mean-field and surface-hopping approaches as both a validative measure to quantitatively assess various electronic structure approaches, as well as to interrogate the potential nonadiabatic contribution to photoreactions of interest, see J. Chem. Phys. 128, 164309 (2008) and Mol. Phys. 108, 1471 (2010).
Water
Despite its ubiquitous presence in our lives, water is surprisingly difficult to computationally simulate to an acceptable level of accuracy. This limits wide-ranging fields including drug design and development of alternative fuels. Computationally tractable approaches require accepting concessions that often regard accounting for molecular polarizibility. Recognizing that the valence bond-based approach applied in previous studies to simulate non-polar solvents naturally incorporates polarizability, we are extending our code base to accommodate simulation of molecules in aqueous environments, see J. Phys. Chem. A 109, 4512 (2005), Chem. Phys. 307, 91 (2004) and J. Chem. Phys. 117, 8867 (2002).